Research
Modeling and Mechanistic Study of Novel Solid Electrolytes
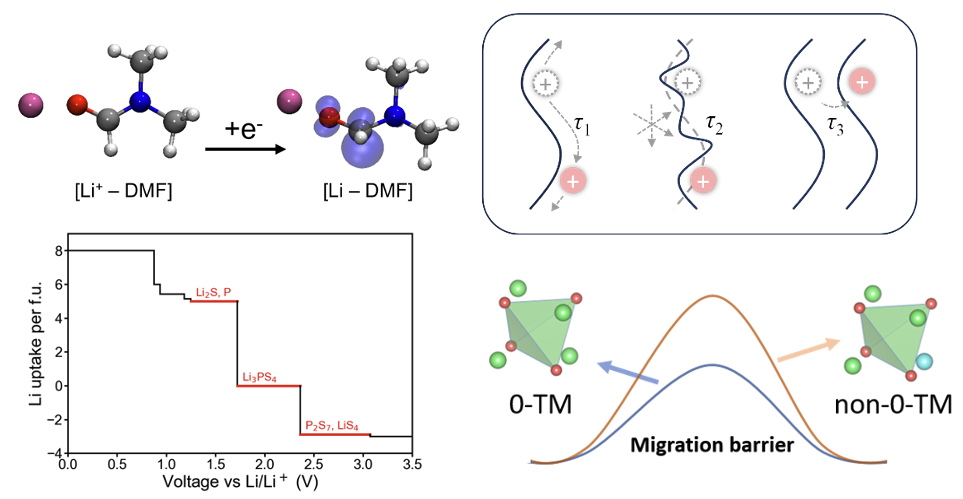
Solid electrolytes (SEs) with high mechanical strength are anticipated to enable the practical implementation of lithium metal batteries and revolutionize the energy storage industry. Compared to conventional liquid electrolytes, they offer enhanced safety, improved low-temperature performance, and higher volumetric energy density. However, their widespread application is hindered by unsatisfactory ion transport and poor interfacial compatability. Traditional experimental techniques struggle to fully characterize atomistic transport and interfacial reaction phenomenon, and comprehensive design principles for novel SEs remain underdeveloped. To address these challenges, our research focuses on the modeling and mechanistic study of SEs, including solid polymer electrolytes, inorganic SEs, and composite SEs. By identifying key structural and chemical factors that govern SE properties, we aim to facilitate the screening and rational design of SEs for practical application.
High-Throughput Computation Infrastructures and High-quality Materials Datasets
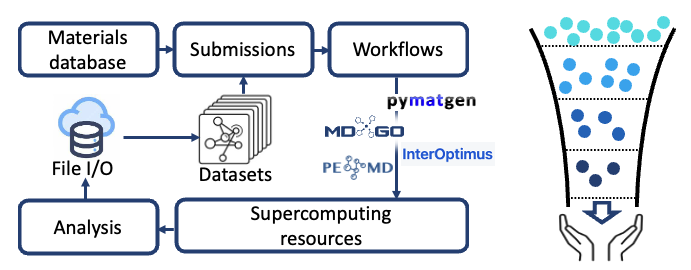
The recent advancement in large language models have spurred the development of AI agents for scientific research. However, natrual language alone cannot fully support the AI-driven materials discovery for SSBs. The major bottleneck remains the lack of high-quality scientific data for training predictive and generative models. To address this challenge, we are developing a high-throughput infrastructure to automate the stability and property calculations of novel SEs. We also leverage advanced universal machine learning interatomic potentials (MLIPs) to accelerate the process after careful benchmarking. Using these tools, we are curating comprehensive property datasets, including ionic conductivity, electrochemical stability windows, electronic structures and mechanical properties. We hope these datasets will facilitate the screening and design of next-generation SE materials. Please stay tuned!
Interface and Interphase in Solid-State Batteries
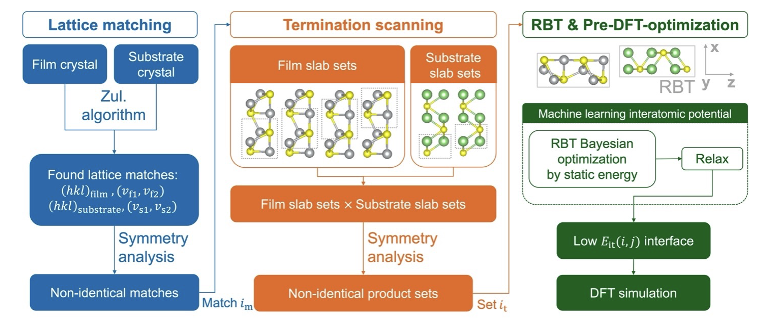
The formation of interphase layers, including the cathode-electrolyte interphase (CEI) and solid-electrolyte interphase (SEI), exhibits significant chemical complexity and plays a pivotal role in determining the performance of SSBs. Despite considerable advances in simulating the bulk phase properties of battery materials, the understanding of interfaces, including crystalline interfaces that represent the simplest case, remains limited. This is primarily due to challenges in performing ground-state searches for interface microstructures and the high computational costs associated with first-principles methods. Therefore, we are currently working on the accurate modeling of the ground-state structures and the structural evolution of interfaces in SSBs. Our downstream investigations involve Li+ transport across interfaces, high-throughput screening for novel artificial interphases, and training predictive and generative models tailored for SSB interfaces.
Predictive and Generative Models for SEs
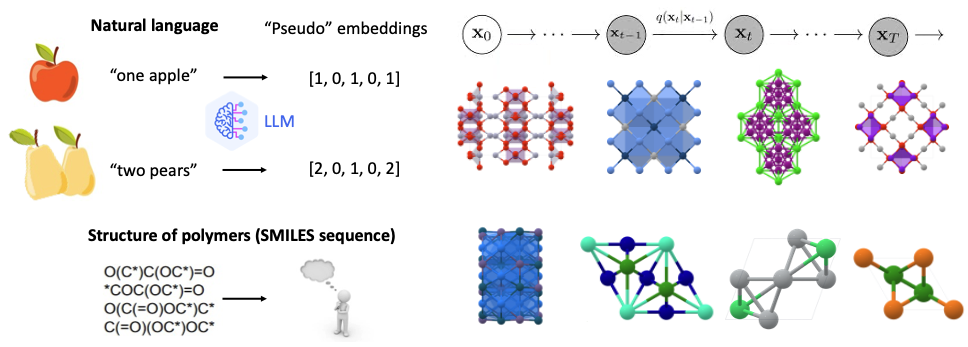
Equiped with the domain expertise and comprehensive datasets, we are developing stability and property prediction models to accelarate the screening of SE materials based on underexplored but important property matriccs. By integrating physics informed modeling with machine-learning techniques, we aim to identify promising SE candidates more efficiently. Additionally, We are utilizing Transformer- and Diffusion-based generative models to facilitate the design of novel SE materials with optimized properties. These AI-driven approaches enable the exploration of a vast structrual and chemical space, generating significantly more candidate materials that are previously unseen. To ensure the reliability and real-world applicability of our predictions, we employ high-accuracy ab initio methods for theoretical validation, followed by experimental synthesis and testing. This iterative approach bridges computational insights with practical implementation, ultimately advancing the discovery and development of next-generation SEs for energy storage applications.